У здорового человека передача нервного импульса осуществляется за счёт мотонейронов передних и задних рогов спинного мозга, которые обеспечивают сократимость клеток и волокон под влиянием медиаторов. В результате подачи сигнала от головного мозга нервный импульс проходит путь через мотонейроны и высвобождается в синапсе, связываясь с рецепторами мышечных волокон, стимулирует их сокращение. С их помощью человеку доступен контроль над движениями, дыханием в организме, функцией мышечной памяти, зависящих от сотрудничества поперечно-полосатых, гладких и сердечных мышц.
У больных со спинально-мышечной атрофией нарушается нервно-мышечная передача, влекущая за собой проблемы с органами дыхания, опорно-двигательным аппаратом, скелетной мускулатурой, мочеполовой системой и сердечно-сосудистой. Иначе говоря, СМА — сочетание заболеваний, возникающих в результате дегенерации двигательных нейронов спинного мозга либо ствола головного, приводящее к снижению мышечного тонуса и силы с дальнейшим развитием пареза или паралича (в зависимости от типа болезни), при этом на умственное развитие и обучаемость заболевания не влияет.

Стоит отметить, чаще характерна спинально мышечная атрофия у детей, но встречается и у взрослых. Частота выявления СМА: 1 новорожденный на 11.000 младенцев.
Тщательное изучение заболевания позволяет придумывать эффективные препараты для лечения при соблюдении этиотропного, симптоматического лечения и медицинской реабилитации.
Однако стоимость фармакологических новшеств снижает их доступность для простого населения.
Причины
В результате исследований генной инженерии патогенез возникновения данного заболевания был раскрыт. Выяснилось, что спинально-мышечная атрофия имеет причины:
- мутацию в геноме SMN1 (наследственная предрасположенность одного из родителей);
- тромбоз кровеносных сосудов;
- родовая травма у младенца;
- перенесённый полиомиелит;
- миозит;
- заболевание поджелудочной железы.
Несмотря на ряд причин, СМА чаще встречается в результате мутации в гене SMN1. Аномалии могут протекать в двух формах: теломерной (SMN1) и центромерной (SMN2).
В первом случае осуществляется замена нуклеотида в 7-м экзоне, что изменяет сплайсинг РНК и нарушает транскрипцию в гене SMN2. Это активирует синтез изменённого разрушающегося белка SMN дельта-7, у которого возникают проблемы с компенсацией делеции SMN1.
Во втором — мутация носит "лёгкий характер", что позволяет синтезировать некоторую часть повреждённого белка SMN. В результате сохраняется частичная функция мотонейронов, ответственных за течение мышечной атрофии. Таким образом, отсутствие "рабочего" гена SMN1 напрямую зависит от SMN2. Копии, выработанные белком N2, не могут восполнить целостность испорченного нуклеотида, что отражается на в форме болезни и возрасте пациента.
Спинальная мышечная атрофия типы
Проксимальная СМА делится на 4 вида, определяющиеся продолжительностью жизни пациента, его возрастом в момент начала болезни и тяжестью течения:
- Младенческий тип (болезнь Верднига-Гофмана). Проявляется болезнь с рождения и в последующие 6 месяцев. Ребёнок имеет проблемы с дыханием (развивается дыхательная недостаточность), нарушен сосательный, кашлевой и глотательный рефлекс (проблемы с проглатыванием слюны и выделением мокроты), мышечная слабость не позволяет держать головку и сидеть. Чтобы продлить жизнь таких пациентов, их подключают к ИВЛ и кормят через зонд. Течение болезни характеризуется задержкой моторного развития, образованием контрактур с деформациями грудной клетки. При должном уходе дети проживают чуть больше двух лет.
- Промежуточный тип (болезнь Дубовица). Проявляется с 7-го по 18 месяц. Ребёнок может самостоятельно принимать пищу, садиться, но не способен передвигаться ногами. Жизнь такого пациента зависит от количества пораженных мышц дыхательного аппарата. При визуальном осмотре наблюдается мышечная скелетная деформация, поверхностное диафрагмальное дыхание, снижение кашлевого рефлекса с переходом в дыхательную недостаточность, контрактуры, тремор конечностей.
- Юношеский тип (болезнь Кюгельберга-Веландера). Болезнь характерна для детей старше полутора лет. До подросткового возраста дети самостоятельно передвигаются, затем происходит утрата моторных навыков, возникают проблемы с глотанием пищи, слюны, пережёвыванием пищи. При осмотре врачом выявляется картина сколиоза, контрактур, прогрессирует дыхательная недостаточность.
- Взрослый тип (взрослая форма). Прогрессирует у лиц старше 35 лет. Пациенты испытывают слабость в мышцах, тремор в конечностях, появляются эндокринологические нарушения, развивается сколиоз, нарушается подвижность в суставах. Первыми начинают отказывать мышцы нижних конечностей, а после — верхних, приводя к сложностям с дыханием и глотанием.
Спинальная мышечная атрофия симптомы

На ранних стадиях больной испытывает усталость, снижение мышечного тонуса, дрожание пальцев рук. В результате перенесённой инфекции поражению подвергаются моторные нейроны. Дегенеративные изменения в них способствуют ограничению движений в коленных, тазобедренных суставах с последующим развитием паралича.
Взрослые и подростки при прогрессировании заболевания во время непродолжительной ходьбы ощущают тяжесть в ногах, онемение в конечностях с трансформацией походки за счёт формирующихся контрактур, мешающих передвижению.
При проявлении дыхательной недостаточности у человека появляется одышка, бледность кожных покровов, синюшность губ (при тяжёлой форме), миофасциальные сокращения, приводящие к боли в результате прикосновений.
У ребенка заподозрить заболевание помогает "поза лягушки" — положение лёжа с разведёнными в сторону ногами, согнутыми в коленных суставах и локтях. При пальпации ягодичной области отмечается снижение тонуса силы, повышенная чувствительность с ощущением боли от прикосновений. Скелетная мускулатура деформируется и формируется сколиоз.
У детей до 2 лет первыми симптомами являются нарушение актов глотания, сосания, откашливания мокроты, невозможность срыгивания (у грудничков), а также обильное слюноотделение и запоры. Во время плача его крик еле слышен из-за регрессивного состояния голосовых связок.
При визуальном осмотре тело худое, суставы имеют контрактуры и тугоподвижность при сгибании-разгибании, грудная клетка деформирована, со спины имеются явные проявления сколиоза. Неврологический осмотр выявляет снижение либо утрату глубоких рефлексов.
Диагностика
Золотым стандартом в определении патологии является метод MLPA (мультикомплексной апплификации лигазно-связанных проб) на стадии планирования беременности и при внутриутробном развитии плода.
В диагностике заболевания помогает осмотр специалистов:
- невролога;
- ортопеда;
- нейрохирурга;
- генетика;
- неонатолога/педиатра;
- эндокринолога и др.
Проводятся лабораторные исследования биохимического анализа крови, где о проблемах с мышечной тканью говорит увеличенная креатинфосфокиназа.
Дополнительные исследования включают:
- Биопсию мышечной ткани. Гистология позволяет определить некротические изменения миофибрилл, увеличение соединительной и разрастание жировой тканей.
- Спирметрию. Оценивает поражение дыхательной системы за счёт функции внешнего дыхания и обструктивных нарушений, снижающих жизненную ёмкость лёгких.
- ЭМГ. Методика обнаруживает участки дегенерации в двигательных нейронах, приводящих к снижению скорости передачи импульса, образованию фасцикуляций с повышенной электрической активностью.
Лечение спинальной мышечной атрофии
После удачного прохождения клинических испытаний в качестве терапии прибегают к лечению СМА препаратами:
- Спинраза;
- Золгенсма.
Каждый из этих препаратов включает введение нескольких нагрузочных доз с промежуточным интервалом, позволить себе данный рецептурные средства может не каждый желающий в силу высокой стоимости. Эффективность препарата замедляет прогрессирование СМА.
Лечением данной патологии занимается невролог, физиотерапевт, нейрохирург.
На каждом этапе используется медикаментозная и немедикаментозная терапия, определяемые тяжестью заболевания.
Использование лекарственных средств направлено на:
- восстановление трофики мышечной ткани;
- синтезирование белка выживаемости мотонейронов;
- предупреждение метаболических нарушений;
- облегчение отхождения мокроты, её разжижение;
- борьбу с гастроэзофагеальным рефлюксом и активации моторики желудочно-кишечного тракта.
Немедикаментозное "восстановление" состоит из физиотерапии и реабилитации. Тандем процедур обеспечивает улучшение моторных функций пациента, укрепляет мышечный корсет и снижает спастичность, но выполняется под надзором специалистов после госпитализации в стационар. К основным мероприятиям, направленным на облегчение его жизнедеятельности относят:
- ЛФК (поддержка тонуса мышечного аппарата путём выполнения активных и пассивных упражнений);
- физиолечение (активирует застоявшиеся метаболические процессы мышечной ткани путём электростимуляции, грязелечения, электрофореза);
- массаж (улучшает действие лимфатической и кроветворной системы, восстанавливая трофику тканей, предотвращая появление пролежней);
- ортопедическая терапия (осуществляется корректировка просто мышечных изменений за счёт использования корсетов, ортопедической обуви).
В тяжёлых формах рекомендовано хирургическое вмешательство и экспериментальное лечение при согласии пациента и его опекунов.
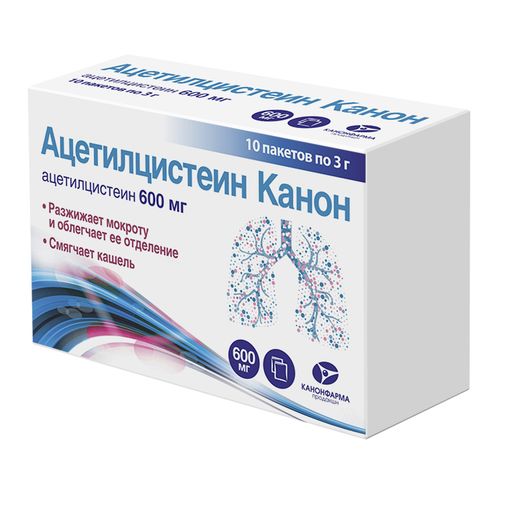
Рекомендованное лекарства
- Ацетилцистеин 200-600 мг.
Порошкообразное муколитическое средство. Активно разжижает сгустки мокроты и облегчают её отделение. - Омепразол 20 мг.
Снижает выработку соляной кислоты и желудочного сока, что позволяет контролировать секрецию слизи. - Коэнзим Q10.
Активирует работу митохондрий, что улучшает обменные процессы в мотонейронах и мышцах. - Итоприд.
Прокинетик. Избавляет пациентов от диспепсии, улучшает моторику ЖКТ.